问题标签 [chemistry]
For questions regarding programming in ECMAScript (JavaScript/JS) and its various dialects/implementations (excluding ActionScript). Note JavaScript is NOT the same as Java! Please include all relevant tags on your question; e.g., [node.js], [jquery], [json], [reactjs], [angular], [ember.js], [vue.js], [typescript], [svelte], etc.
neural-network - 可以使用 Java 通过 ANN 对量子力学行为进行建模吗?
我正在做一个独立的项目。我在学校学习化学和计算机科学,想知道是否可以使用人工神经网络对某些波函数现象(薛定谔方程、哈密顿方程、特征值)进行建模。
我的主要问题是:
- 我可以在笔记本电脑上编程和计算吗?我的笔记本电脑是华硕 Q200e
- 如果不能从笔记本电脑上使用,我是否可以使用包含 i5 处理器和快速 GPU 的台式机?
lua - Lua中的分子量计算器
作为我在 Lua 中的第一个真正的程序,我想创建一个分子量计算器(类似于这里的那个:http: //www.lenntech.com/calculators/molecular/molecular-weight-calculator.htm)
我想在这个程序的第一步得到一些帮助。我的第一步应该是将用户输入作为字符串,而不是拆分字符串。我正在查看http://lua-users.org/wiki/StringLibraryTutorial并试图弄清楚如何做到这一点。当用户输入为 CH4 时,程序应将此字符串拆分为C, 和H4。这是我的代码:
有人可以告诉我如何接受用户输入作为字符串以及如何拆分该字符串吗?
编辑:根据要求,我在我的确切问题中更加具体
regex - 替换化学式字符串中的文本
我无法编写一个函数,该函数将采用诸如“NiNFe(AsO2)2”之类的化学式字符串并删除其中一个元素。
我目前的尝试是:
如果符号为“N”且公式为“NiNFe(AsO2)2”,我将得到“iFe(AsO2)2”而不是所需的“NiFe(AsO2)2”。有谁知道如何以这样的方式对其进行编码,以便将 N 与 Ni 区分开来并替换它?
python - 求解给定变量和不确定性的线性方程:scipy-optimize?
我想最小化一组方程,其中变量的不确定性是已知的。本质上,我想检验给定测量变量符合方程给出的公式约束的假设。这似乎是我应该能够用 scipy-optimize 做的事情。例如我有三个方程:
以及四个具有 1-sigma 不确定性的测量未知数:
寻找正确方向的任何指示。
python - 构建蛋白质数据库 (PDB) 文件
我必须模拟一个复杂的赖氨酸聚合物。它类似于蛋白质,但赖氨酸并不总是与它们的 α 胺结合。我的目标是生成一个PDB(蛋白质数据库)以供进一步计算。
你知道任何允许我构建分子的模块吗?我有特殊需求,例如:
- 我有氨基酸链
- 我需要在链中再添加一个
- 但我必须能够指定这个 n+1 氨基酸在哪里以及如何与前一个氨基酸结合
- 最后,我必须能够生成 PDB 文件
我的第一种方法是使用 SMILES 格式,我可以完美地完成所有操作,除了最后无法构建 pdb,因为没有软件能够处理我的原子数(> 15000)。
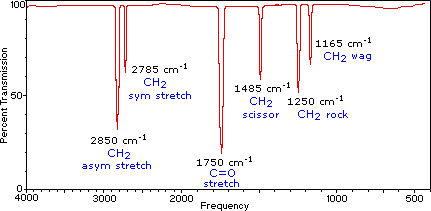
plot - 使用 Gnuplot 绘制红外光谱
我有一个想要绘制的感兴趣化合物的红外光谱,并且我有一个包含所有数据点的spectrum.dat文件。它的形式是:
我想使用典型的 IR 光谱的 x 轴来绘制此图,但我在这样做时遇到了麻烦。如果您不熟悉,这就是典型的 IR 光谱的样子(除了图表本身的标签)。请注意,x 轴是反转的,并且在超过 2000 个单位(厘米倒数)时,它的缩放比例突然加倍。有没有办法强迫 Gnuplot 以这种方式绘制我的数据?到目前为止,我已经设法想出了以下脚本:
{kind=link}
这很好地反转了 x 轴,但我无法弄清楚如何以这种不寻常的方式更改缩放比例。
r - ChemoSpec 找不到 getManyCsv 函数
我正在尝试创建 Spectra 对象 (.RData),就像在 ChemoSpec.pdf 上建议的那样(参见下面的代码),我收到如下错误消息:
“找不到 getManyCsv 函数”
我该如何解决?任何建议表示赞赏。我有 R 版本 3.1.0 和 ChemoSpec 版本 2.0-2。
这是代码:
干杯,
阿维尔
javascript - Javascript ROY G BIV 线谱?
我对 javascript 相当陌生,我想使用 html canvas 和 javascript 制作一个小线谱,以便以后可以在单击按钮时对其进行动画处理以显示吸收线等,就像在化学中一样。我只是不确定纯javascript是否能够处理这个问题。谁能提供一些关于如何做到这一点的信息,或者这是否可能?如果需要,我很乐意提供更多信息。
编辑:这是我到目前为止所拥有的,但我不知道为什么颜色渐变会在颜色变化的地方倾斜,并且需要调整颜色的宽度,以便它们沿矩形占据相等的数量。
HTML
JavaScript
}
loadSpectralCanvas();
r - R:基于自定义距离函数和多个条件快速匹配记录
我在 R 中创建了一些函数,以根据自定义光谱相似性函数和所谓的化合物保留指数匹配(即洗脱时间)(参见此处的示例,http: //webbook.nist.gov/cgi/cbook.cgi?ID=C630035&Mask=200)。为此,必须将每个记录的列表元素“RI”与库中的元素进行比较,当绝对偏差小于给定容差时,它应该将最佳光谱库匹配添加到我的记录中。下面是我为此编写的一些代码,但问题是它对于我的目的来说太慢了(我通常有大约 1000 个样本光谱和 200 000 个库光谱)。我尝试并行化它,但这似乎也没有多大帮助。关于如何使下面的代码更高效的任何想法,例如使用更多矢量化,或使用内联 C 代码,或其他一些 R 技巧?我知道这方面的一般建议,但不太明白在这种情况下如何轻松实现它(不幸的是,我对 C 语言还不是很精通)......有什么想法或建议吗?哦是的,sfLapply? 首先将我的光谱放在一个大(稀疏,因为有很多零)矩阵中是否有帮助,以避免merge光谱相似性函数中的步骤,或者使用其他标准,例如仅在最大/最多时考虑光谱查询光谱中的强峰与库光谱具有相同的质量(或者包含在库光谱中的 5 个最大峰的集合中)?无论如何,任何关于如何加快这项任务的想法都将不胜感激!
编辑:我仍然有一个剩余查询是如何避免在函数 addbestlibmatches1 中制作样本记录 recs 的完整副本,而是只更改存在库匹配的记录?此外,传递保留索引匹配的图书馆记录的选择可能效率不高(在函数 addbestlibmatch 中)。有什么想法可以避免这种情况吗?