我被一个问题困扰了三天......到处搜索,发布在Biostar上,仍在等待 EMBL 回复电子邮件......如果我有更多的代表,将会获得赏金。
在使用 EMBOSSwin needle()(成对全局对齐)对齐序列后,我得到pair格式的对齐文件,带有.needle文件扩展名。我想使用Biopython读取这些对齐方式以供以后分析。
我AlignIO.read(open('alignment.needle'),'emboss')按照Biopython 的 AlignIO wiki中的说明使用,但我不断得到一个AssertionError.
我的代码:
>>> from Bio import AlignIO
>>> alignment = AlignIO.read(open("data/all/out/pair1_alignment.needle"), "emboss")
我的错误:
Traceback (most recent call last):
File "<console>", line 1, in <module>
File "C:\Python27\lib\Bio\AlignIO\__init__.py", line 423, in read
first = next(iterator)
File "C:\Python27\lib\Bio\AlignIO\__init__.py", line 370, in parse
for a in i:
File "C:\Python27\lib\Bio\AlignIO\EmbossIO.py", line 150, in __next__
assert seq.replace("-", "") != ""
AssertionError
对齐文件示例:
在此处下载对齐文件
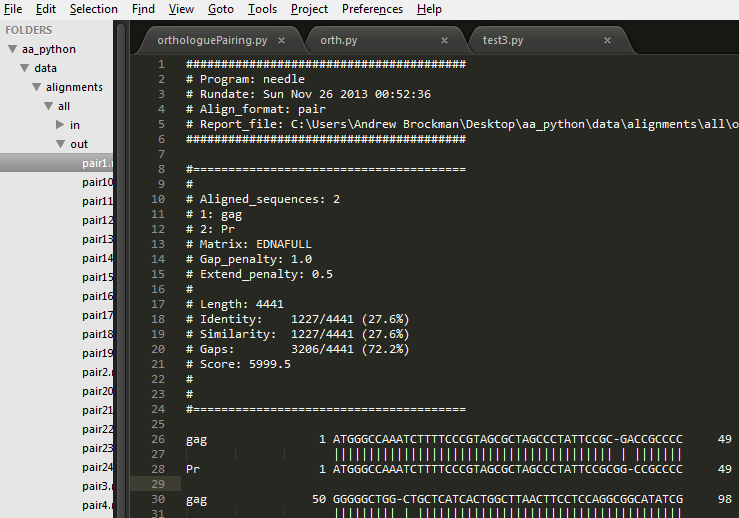
版本:
- Windows 7的
- Python 版本 2.7.3
- Biopython 1.63 版
- EMBOSS 版本 2.10.0-0.8
线索:
我怀疑这可能与我在实际进行对齐时不断收到的警告消息有关,该消息由 EMBOSSneedle()函数输出:
Warning: Sequence character string not found in ajSeqCvtKS